I. Charge transport in electronic materials
Carrier scatterings with phonons and traps are the key factors in evaluating the mobility. We consider three major scattering regimes:
-
- where the molecular internal vibration severely induces charge self-trapping and, thus, the hopping mechanism dominates;
- where both intermolecular and intramolecular scatterings come to play roles, so the Holstein-Peierls polaron model is applied;
- where charge is well delocalized with coherence length comparable with acoustic phonon wavelength, so that a deformation potential approach is more appropriate.
We develop computational methods at the first-principles level for the three different cases that have extensive potential application in rationalizing material design.
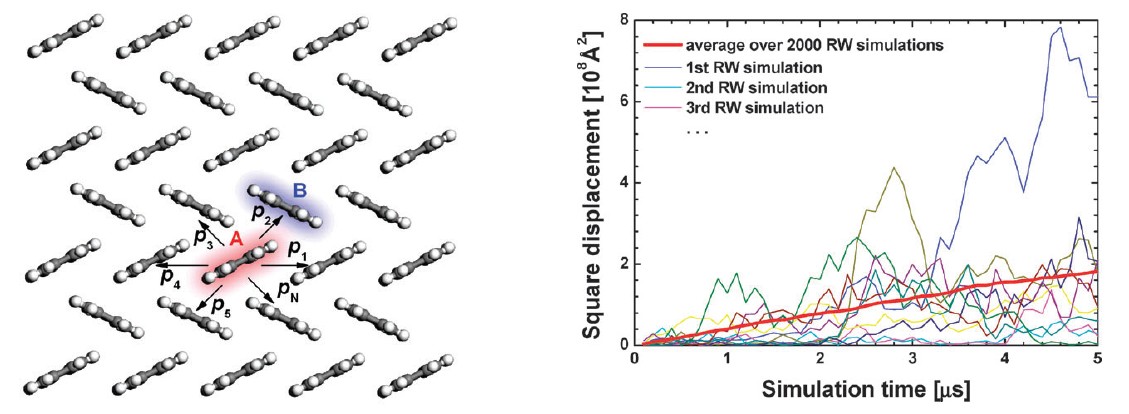
Schematic representation of charge hopping pathway and the typical results of Monte Carlo random walk
Adv. Mater., 2011, 23 (9), 1145-1153 Chem. Soc. Rev. 2010, 39, 423-434
II. Excited states and light-emitting materials
General formalism of absorption and emission spectra, and of radiative and nonradiative decay rates are derived by using a thermal vibration correlation function formalism for the transition between two adiabatic electronic states in polyatomic molecules. Displacements, distortions, and Duschinsky rotation of potential energy surfaces are included within the framework of a multidimensional harmonic oscillator model. The Herzberg−Teller (HT) effect is also taken into account.
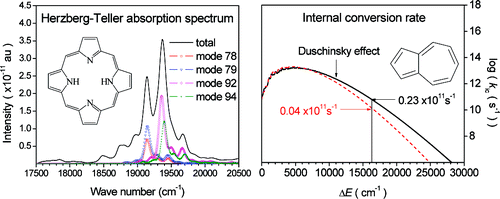
J. Phys. Chem. A, 2010, 114 (30), 7817–7831 J. Am. Chem. Soc. 2007, 129(30), 9333-9339
III. Solar cells modelings
We applied the theoretical models and computational tools in assessing the performance of organic solar cells, especially the bulk heterojunction solar cells. Both the continuum device model and the dynamic Monte Carlo model are developed to investigate the photocurrent-voltage characteristics based on the exciton and charge carrier dynamics.
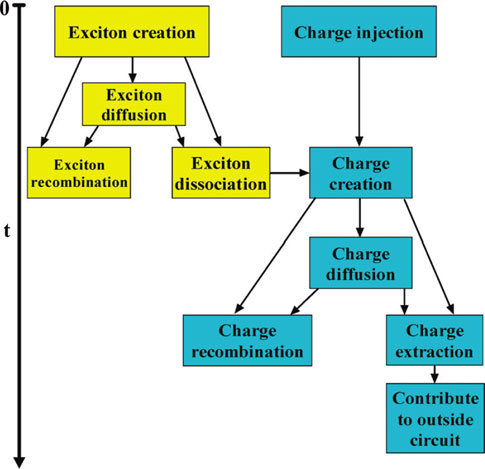
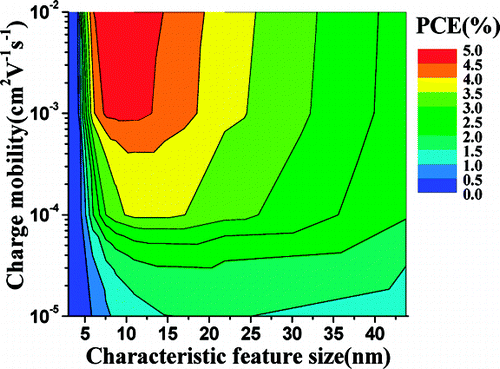
J. Phys. Chem. B, 2010, 114, 36-41 Theor. Chem. Acc. 2011, 129(3-5), 291-301
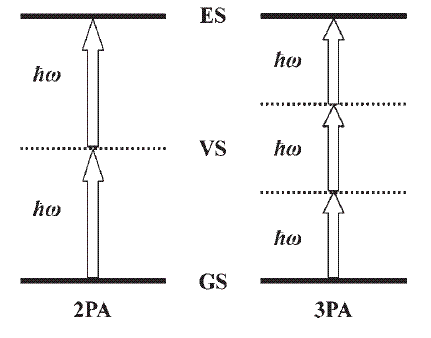
IV. Electron correlation and nonlinear optical responses
The influences of intrinsic molecular structure parameters on the multiphoton absorption properties for organic conjugated molecules include the following terms:
-
- the donor/acceptor substitutions;
- the π conjugation length;
- the ground-state polarization mimicking structural character such as bond-length alternation;
- the molecular dimensionality;
- some other possible factors, such as vibronic coupling and the solvent, as well as aggregation effects.
Some theoretical designing strategies for organic materials with large multiphoton cross-sections are proposed.
Macromol. Theory Simul., 2008, 17, 12-22
For more detailed information on our work, please check our Publications page or contact us directly.